# import sample labels
samples <- read.delim("../../../Reg_Evo_Primates/data/Sample_info_RNAseq_RIN.txt")
RIN <- samples$RIN
# expression
exprs <- read.table("../../../Reg_Evo_Primates/data/250_exp_avg_methyl_hcr_4155_genes.txt", sep="")
# methylation data
methyl <- read.csv("../../../Reg_Evo_Primates/data/250_avg_methyl_hcr_4155_genes_unmerged.txt", sep="")
# Normalized gene expression data
cpm.voom.cyclic <- readRDS("../../../Reg_Evo_Primates/data/human_chimp_orth_cpm_voom_cyclic.rds")
results <- readRDS("../data/combined-limma.rds")
# Load libraries
library("edgeR")
## Loading required package: limma
library("limma")
library("plyr")
library("ashr")
library("cowplot")
## Warning: package 'cowplot' was built under R version 3.4.4
## Loading required package: ggplot2
## Warning: package 'ggplot2' was built under R version 3.4.4
##
## Attaching package: 'cowplot'
## The following object is masked from 'package:ggplot2':
##
## ggsave
library("devtools")
## Warning: package 'devtools' was built under R version 3.4.4
library("vashr")
## Loading required package: SQUAREM
## Loading required package: qvalue
devtools::install_github("jhsiao999/mediation/pkg")
## Skipping install of 'medinome' from a github remote, the SHA1 (dfd53a1b) has not changed since last install.
## Use `force = TRUE` to force installation
library("medinome")
Run the linear model (FDR level = 0.01)
human_rhesus_heart <- c(17:31)
FDR_level <- 0.01
X <- c(0,0,0,1,0,0,0,1,0,0,0,1,0,0,0)
RIN_subset <- RIN[human_rhesus_heart]
# Run for hearts
# Run the linear model in limma
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Heart_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Heart_fit_all_5perc <-
HvC_Heart_fit_all[which(HvC_Heart_fit_all$adj.P.Val < FDR_level), ]
# Run for kidneys
X <- c(1,0,0,0,1,0,0,0,1,0,0,0,1,0,0)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Kidney_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Kidney_fit_all_5perc <-
HvC_Kidney_fit_all[which(HvC_Kidney_fit_all$adj.P.Val < FDR_level), ]
# Run for livers
X <- c(0,1,0,0,0,1,0,0,0,1,0,0,0,1,0)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Liver_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Liver_fit_all_5perc <-
HvC_Liver_fit_all[which(HvC_Liver_fit_all$adj.P.Val < FDR_level), ]
# Run for lungs
X <- c(0,0,1,0,0,0,1,0,0,0,1,0,0,0,1)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Lung_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Lung_fit_all_5perc <- HvC_Lung_fit_all[which(HvC_Lung_fit_all$adj.P.Val < FDR_level), ]
dim(HvC_Heart_fit_all_5perc)
## [1] 628 7
dim(HvC_Kidney_fit_all_5perc)
## [1] 317 7
dim(HvC_Liver_fit_all_5perc)
## [1] 906 7
dim(HvC_Lung_fit_all_5perc)
## [1] 141 7
Heart specific
heart_spec <- union(union(rownames(HvC_Kidney_fit_all_5perc), rownames(HvC_Liver_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Heart_fit_all_5perc$genes, heart_spec)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 205 31
dim(methylation_values_only)
## [1] 205 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
X <- c(0,0,0,1,0,0,0,1,0,0,0,1,0,0,0)
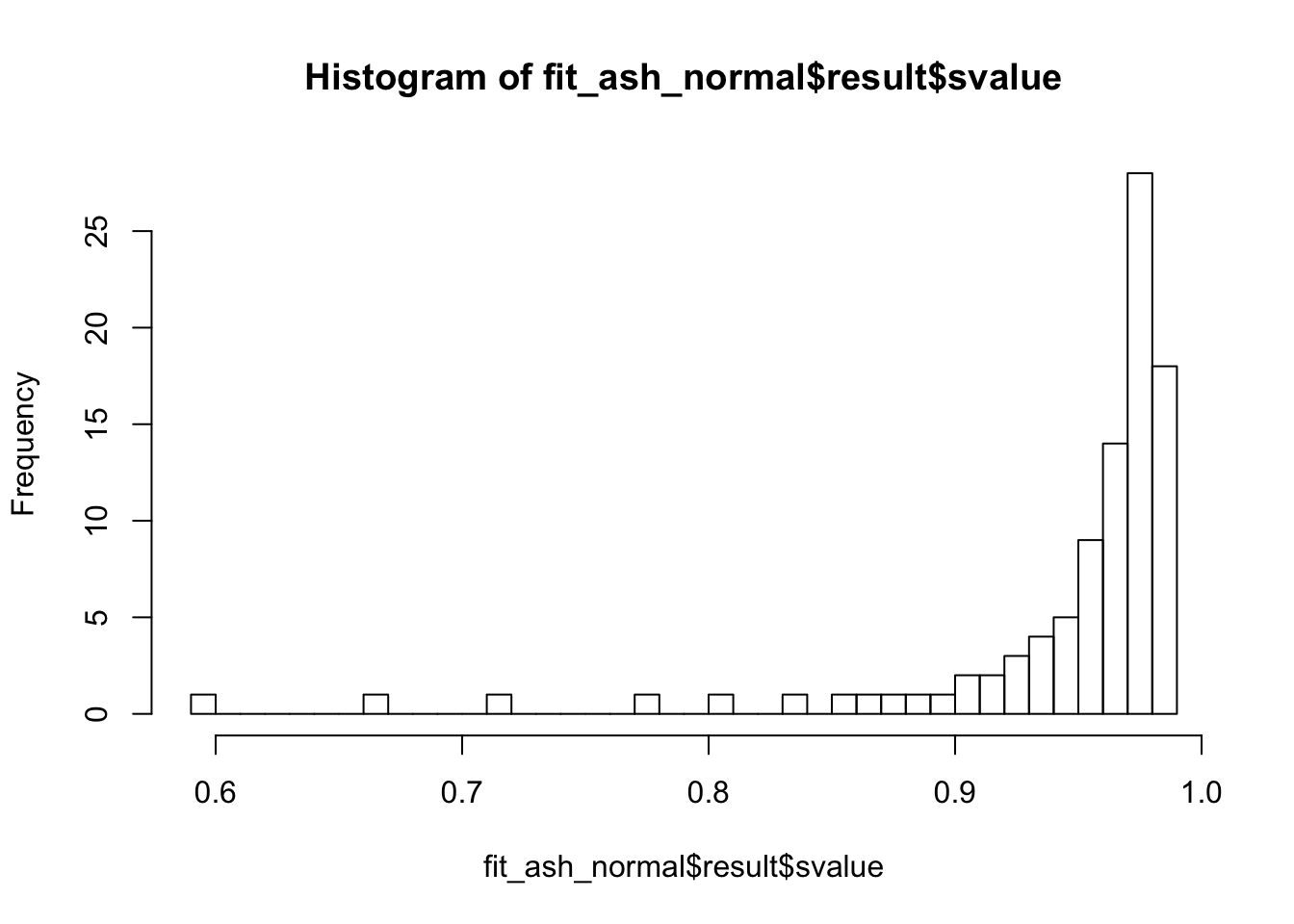
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
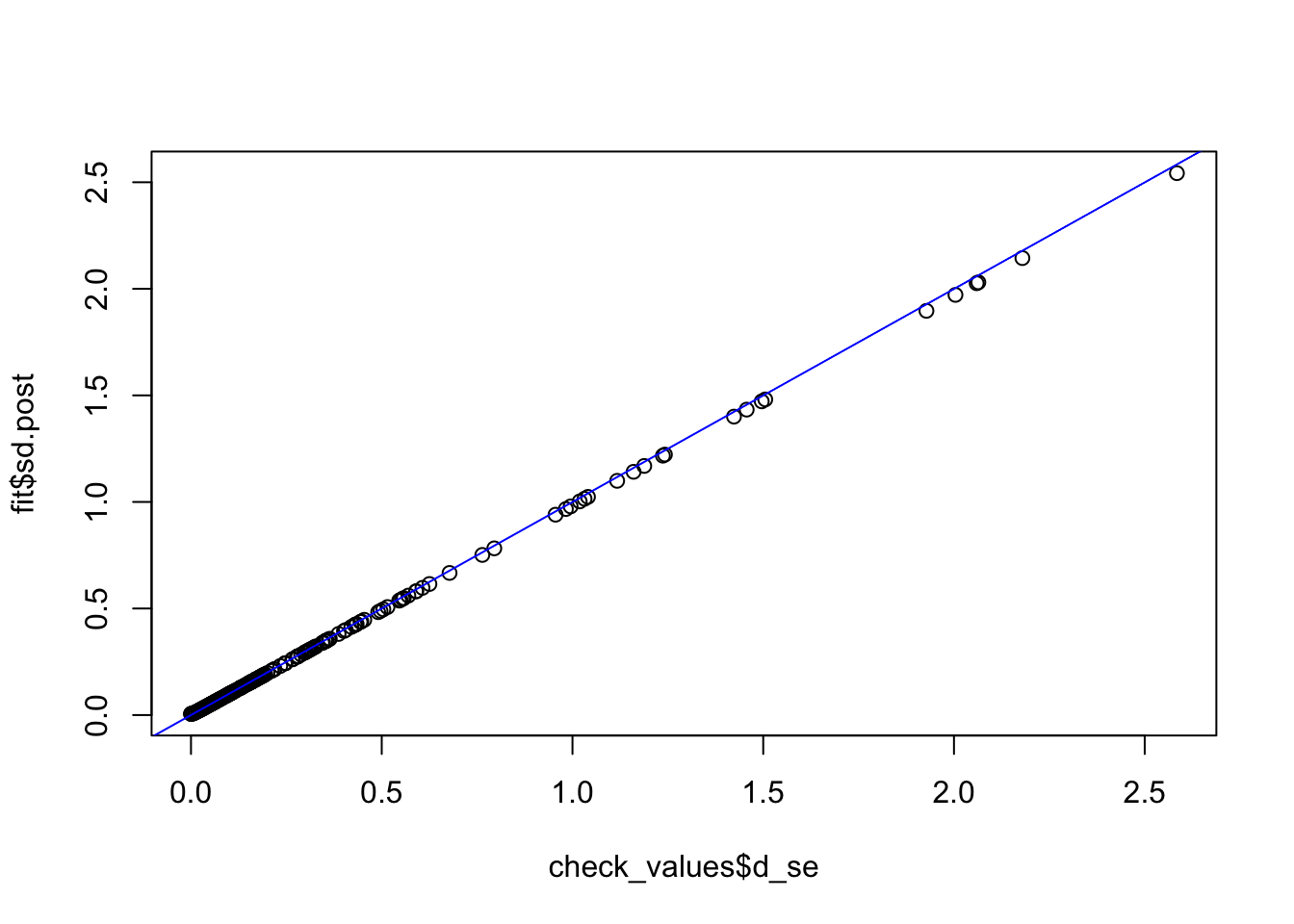
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE
## logical 205
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE
## logical 205
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 202 3
hist(fit_ash_normal$result$svalue, breaks = 50)
#which(fit_ash_normal$result$svalue < 0.1 = TRUE)
Kidney specific
heart_spec <- union(union(rownames(HvC_Heart_fit_all_5perc), rownames(HvC_Liver_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Kidney_fit_all_5perc$genes, heart_spec)
X <- c(1,0,0,0,1,0,0,0,1,0,0,0,1,0,0)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 92 31
dim(methylation_values_only)
## [1] 92 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
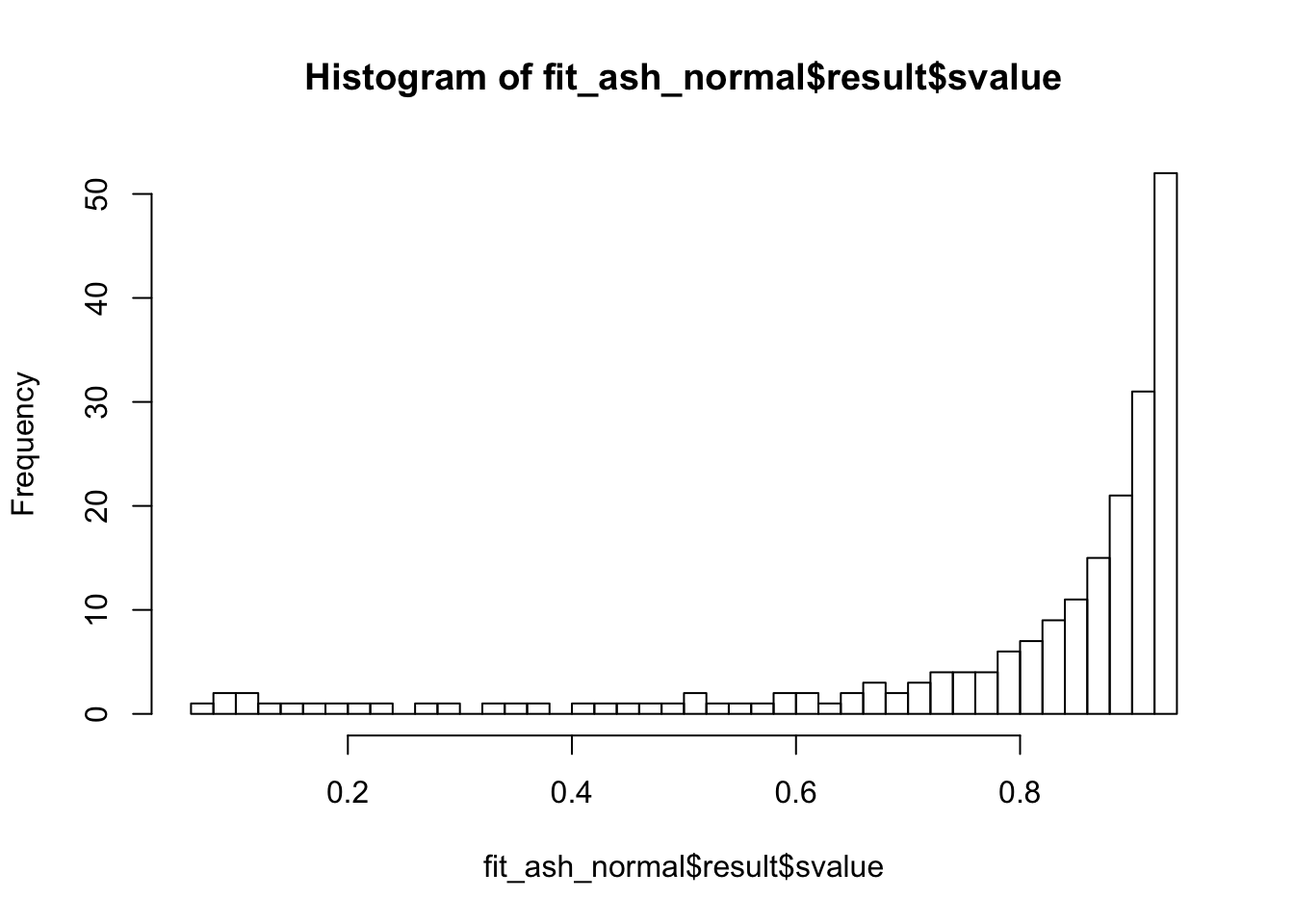
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
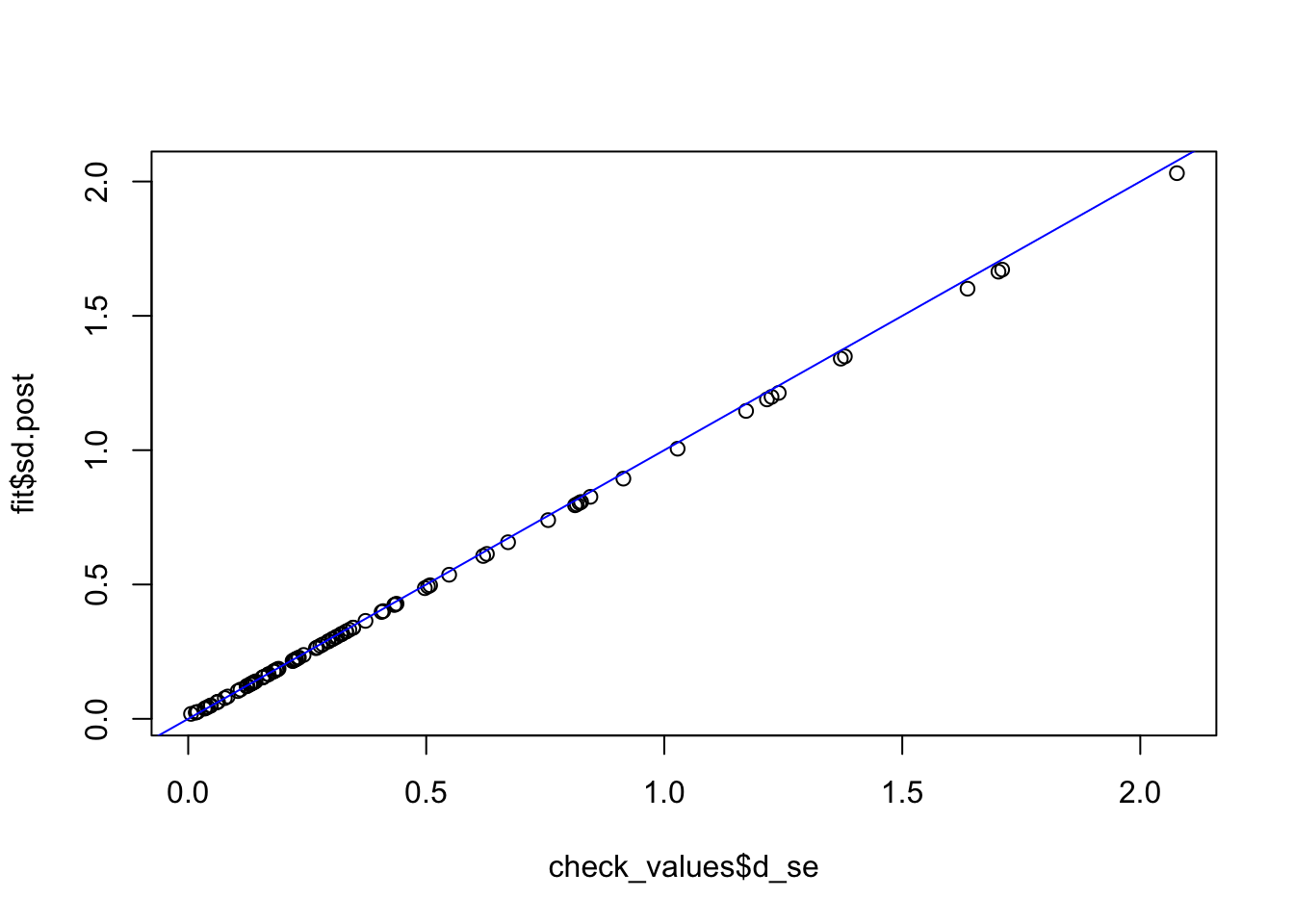
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE TRUE
## logical 88 4
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE TRUE
## logical 87 5
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 86 6
hist(fit_ash_normal$result$svalue, breaks = 50)
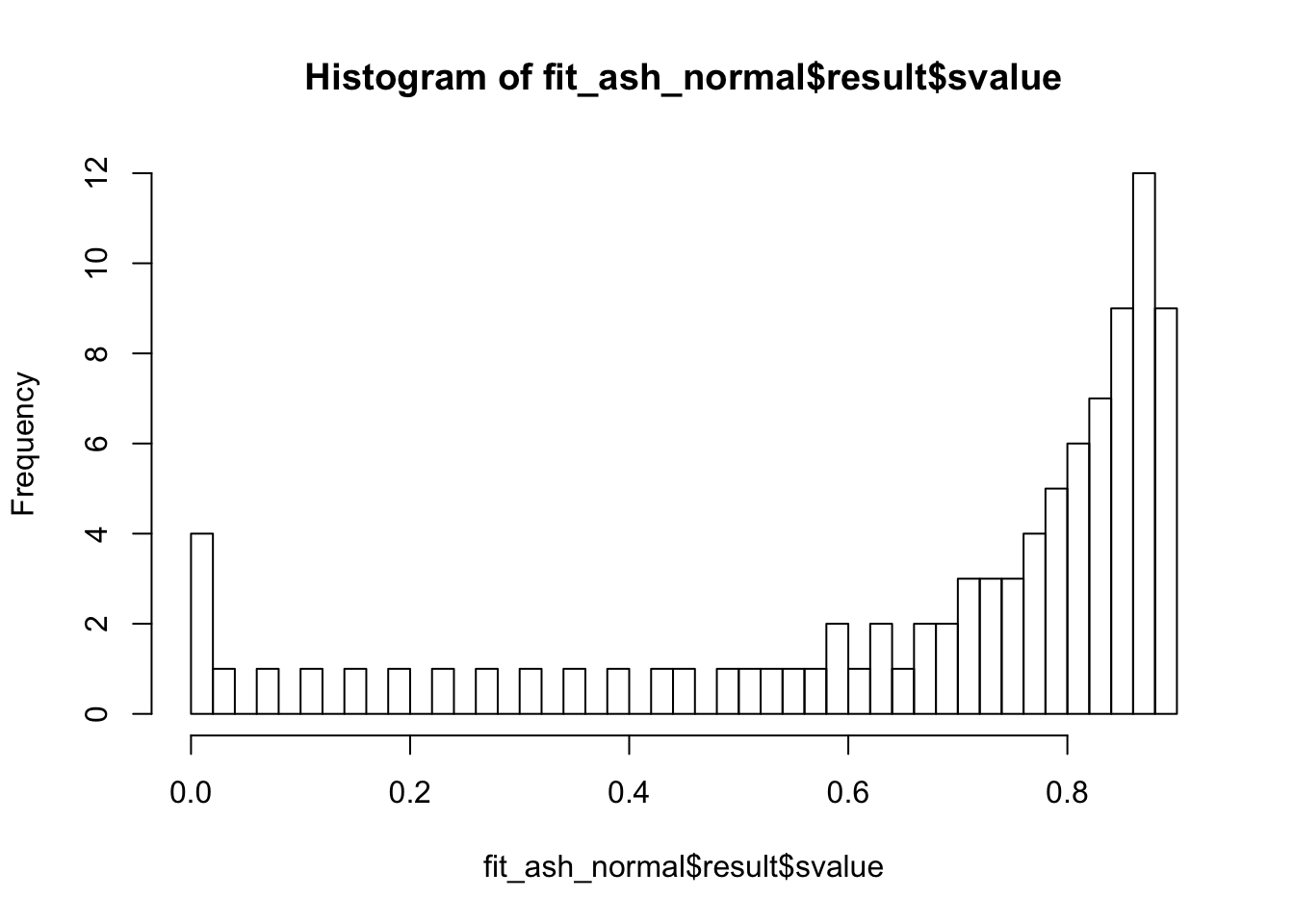
Liver specific
heart_spec <- union(union(rownames(HvC_Heart_fit_all_5perc), rownames(HvC_Kidney_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Liver_fit_all_5perc$genes, heart_spec)
X <- c(0,1,0,0,0,1,0,0,0,1,0,0,0,1,0)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 261 31
dim(methylation_values_only)
## [1] 261 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
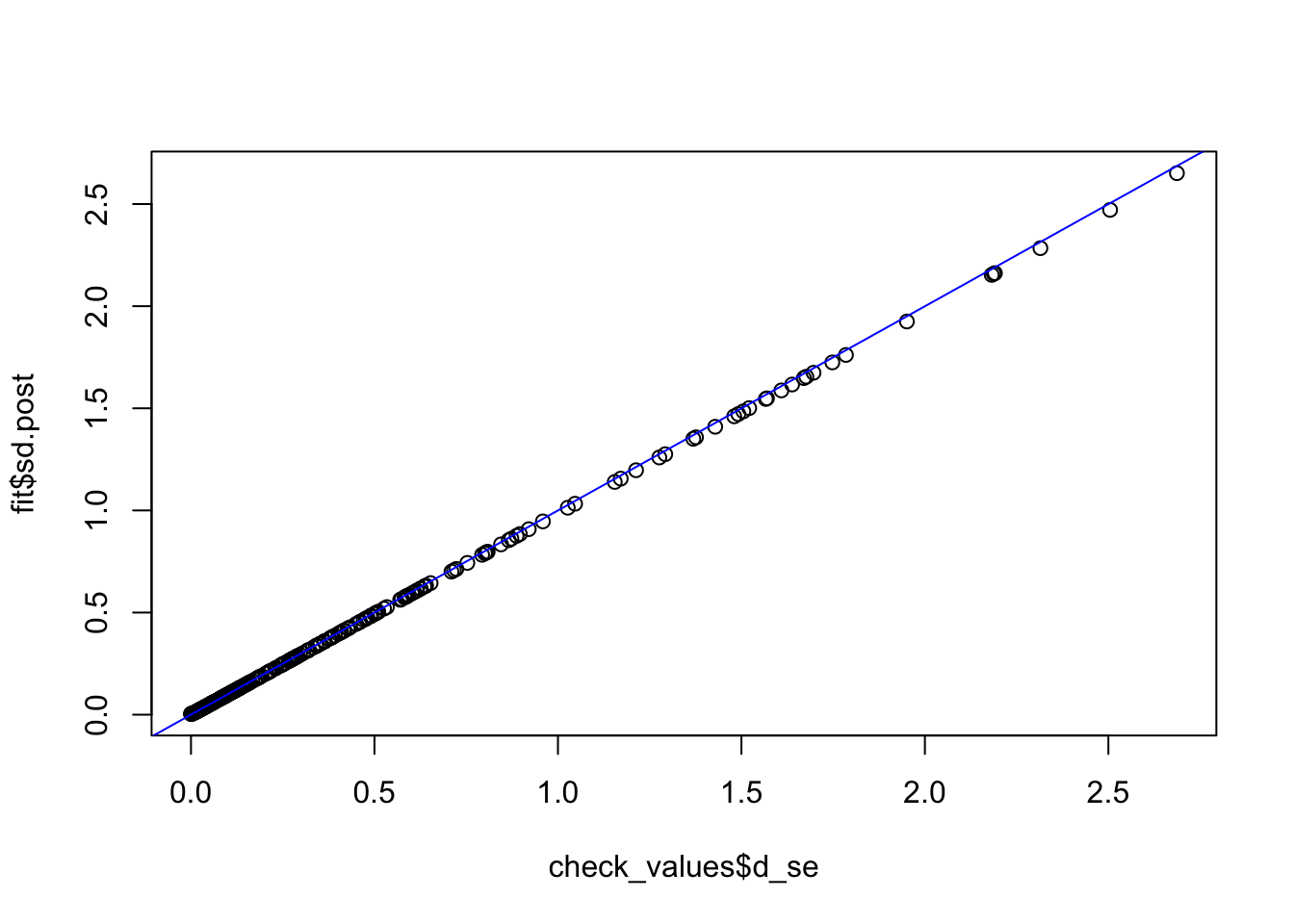
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE TRUE
## logical 257 4
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE TRUE
## logical 253 8
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 250 11
hist(fit_ash_normal$result$svalue, breaks = 50)
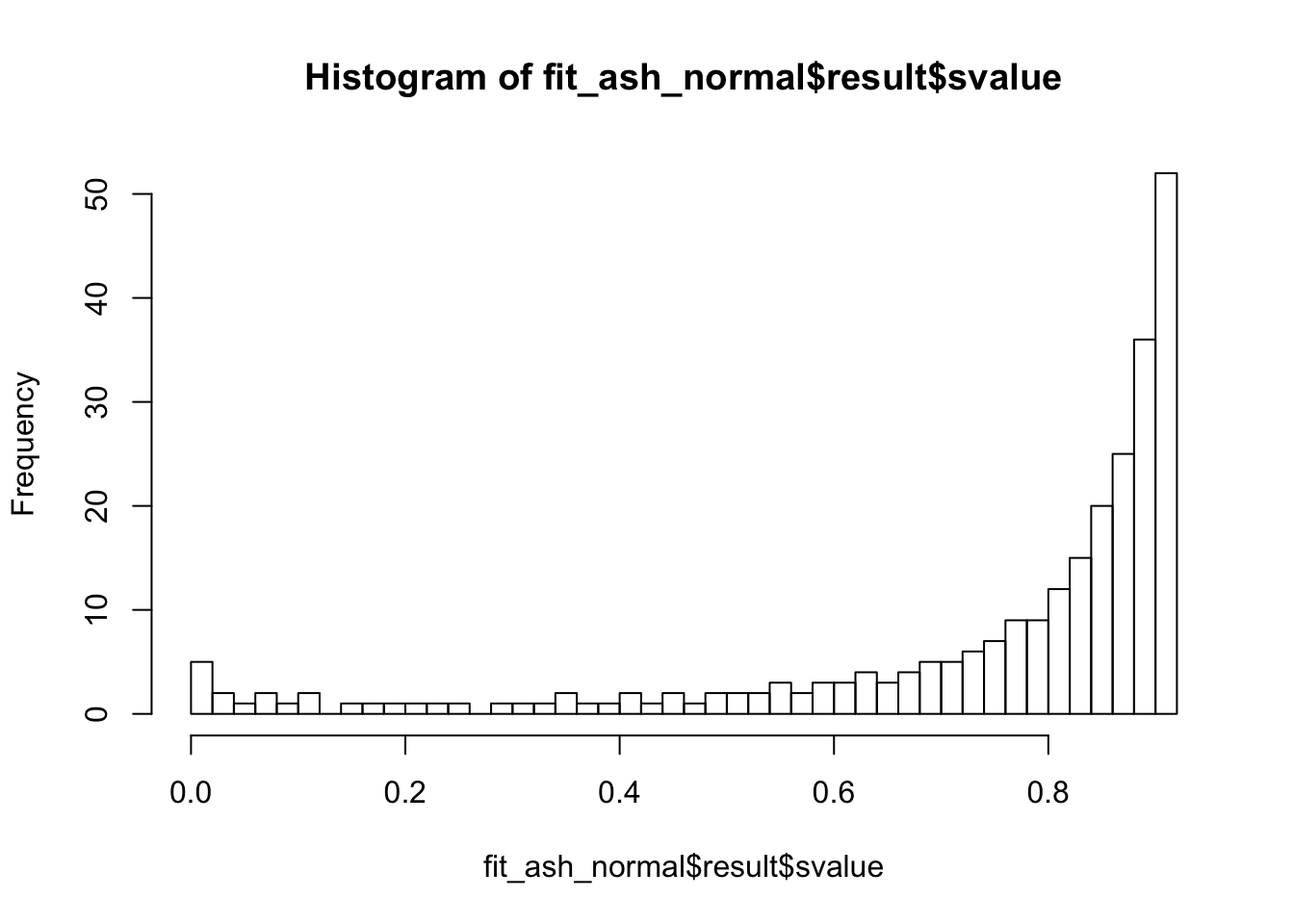
Lung specific
heart_spec <- union(union(rownames(HvC_Liver_fit_all_5perc), rownames(HvC_Heart_fit_all_5perc)), rownames(HvC_Kidney_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Lung_fit_all_5perc$genes, heart_spec_only)
X <- c(0,0,1,0,0,0,1,0,0,0,1,0,0,0,1)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 48 31
dim(methylation_values_only)
## [1] 48 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
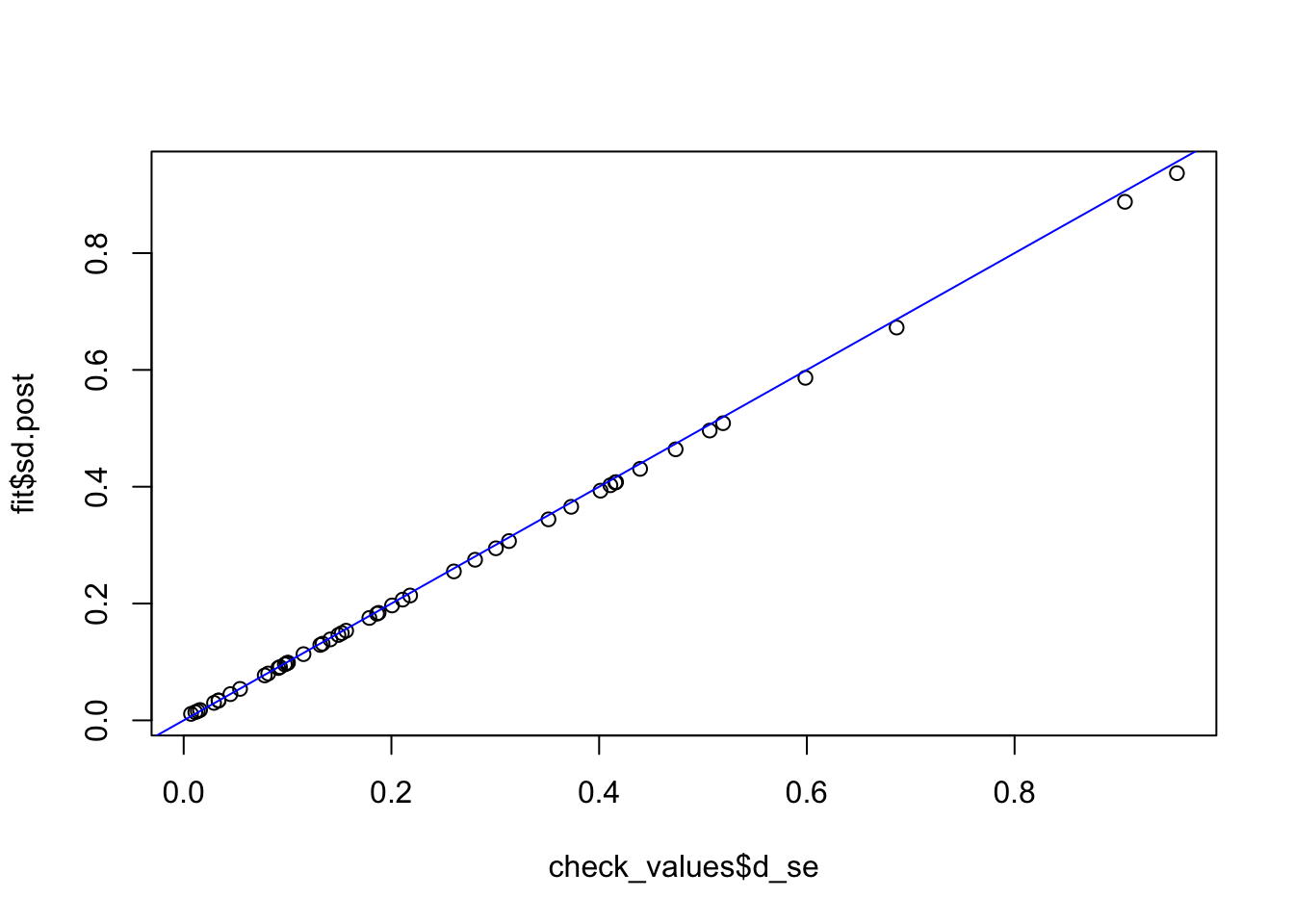
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE
## logical 48
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE
## logical 48
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE
## logical 48
hist(fit_ash_normal$result$svalue, breaks = 50)
Run the linear model (FDR level = 0.05)
human_rhesus_heart <- c(17:31)
FDR_level <- 0.05
X <- c(0,0,0,1,0,0,0,1,0,0,0,1,0,0,0)
RIN_subset <- RIN[human_rhesus_heart]
# Run for hearts
# Run the linear model in limma
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Heart_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Heart_fit_all_5perc <-
HvC_Heart_fit_all[which(HvC_Heart_fit_all$adj.P.Val < FDR_level), ]
# Run for kidneys
X <- c(1,0,0,0,1,0,0,0,1,0,0,0,1,0,0)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Kidney_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Kidney_fit_all_5perc <-
HvC_Kidney_fit_all[which(HvC_Kidney_fit_all$adj.P.Val < FDR_level), ]
# Run for livers
X <- c(0,1,0,0,0,1,0,0,0,1,0,0,0,1,0)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Liver_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Liver_fit_all_5perc <-
HvC_Liver_fit_all[which(HvC_Liver_fit_all$adj.P.Val < FDR_level), ]
# Run for lungs
X <- c(0,0,1,0,0,0,1,0,0,0,1,0,0,0,1)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Lung_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Lung_fit_all_5perc <- HvC_Lung_fit_all[which(HvC_Lung_fit_all$adj.P.Val < FDR_level), ]
dim(HvC_Heart_fit_all_5perc)
## [1] 1281 7
dim(HvC_Kidney_fit_all_5perc)
## [1] 848 7
dim(HvC_Liver_fit_all_5perc)
## [1] 1953 7
dim(HvC_Lung_fit_all_5perc)
## [1] 838 7
Heart specific
heart_spec <- union(union(rownames(HvC_Kidney_fit_all_5perc), rownames(HvC_Liver_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Heart_fit_all_5perc$genes, heart_spec)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 362 31
dim(methylation_values_only)
## [1] 362 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
X <- c(0,0,0,1,0,0,0,1,0,0,0,1,0,0,0)
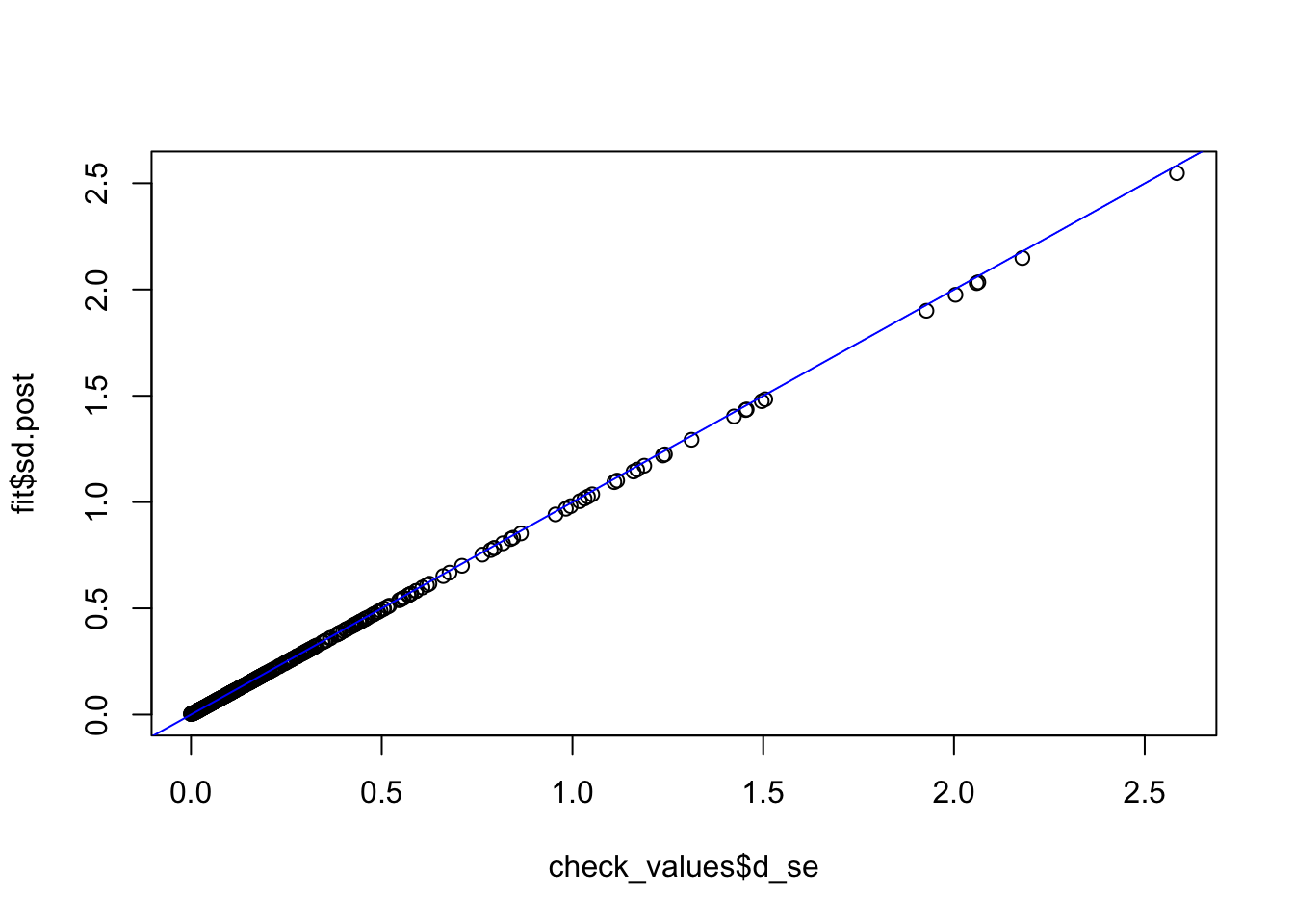
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
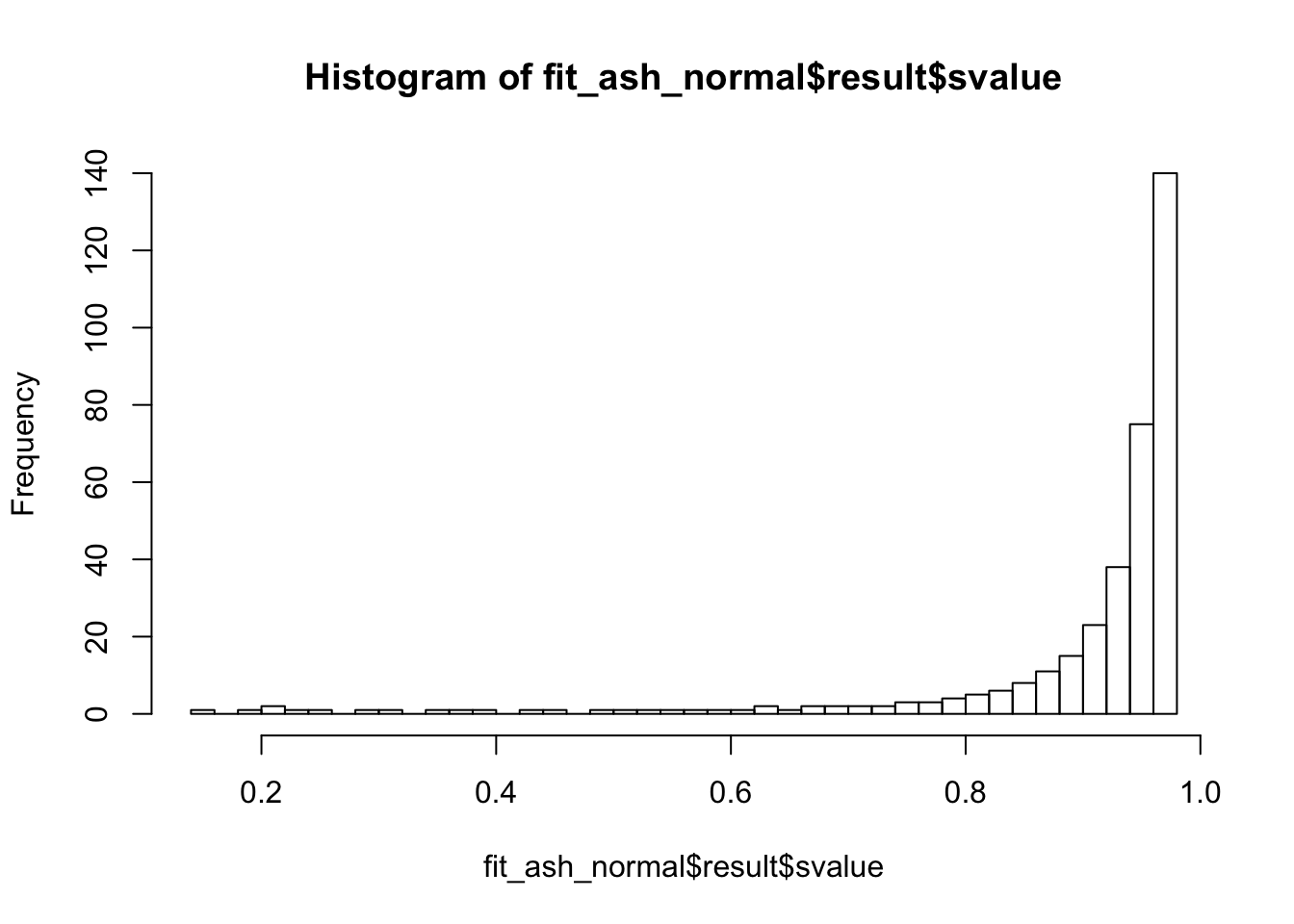
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE
## logical 362
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE
## logical 362
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE
## logical 362
hist(fit_ash_normal$result$svalue, breaks = 50)
#which(fit_ash_normal$result$svalue < 0.1 = TRUE)
Kidney specific
heart_spec <- union(union(rownames(HvC_Heart_fit_all_5perc), rownames(HvC_Liver_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Kidney_fit_all_5perc$genes, heart_spec)
X <- c(1,0,0,0,1,0,0,0,1,0,0,0,1,0,0)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 176 31
dim(methylation_values_only)
## [1] 176 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
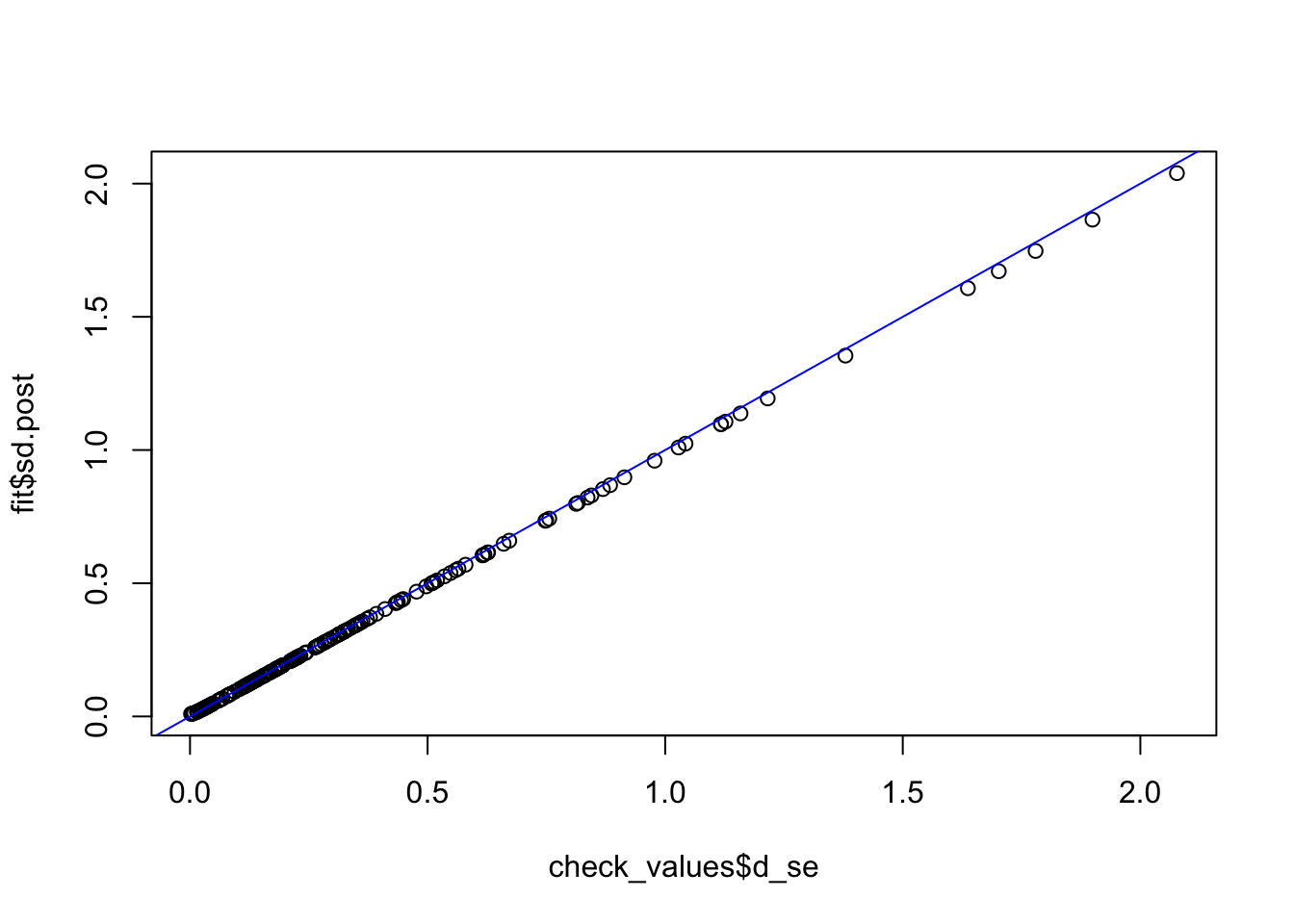
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE TRUE
## logical 173 3
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE TRUE
## logical 172 4
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 171 5
hist(fit_ash_normal$result$svalue, breaks = 50)
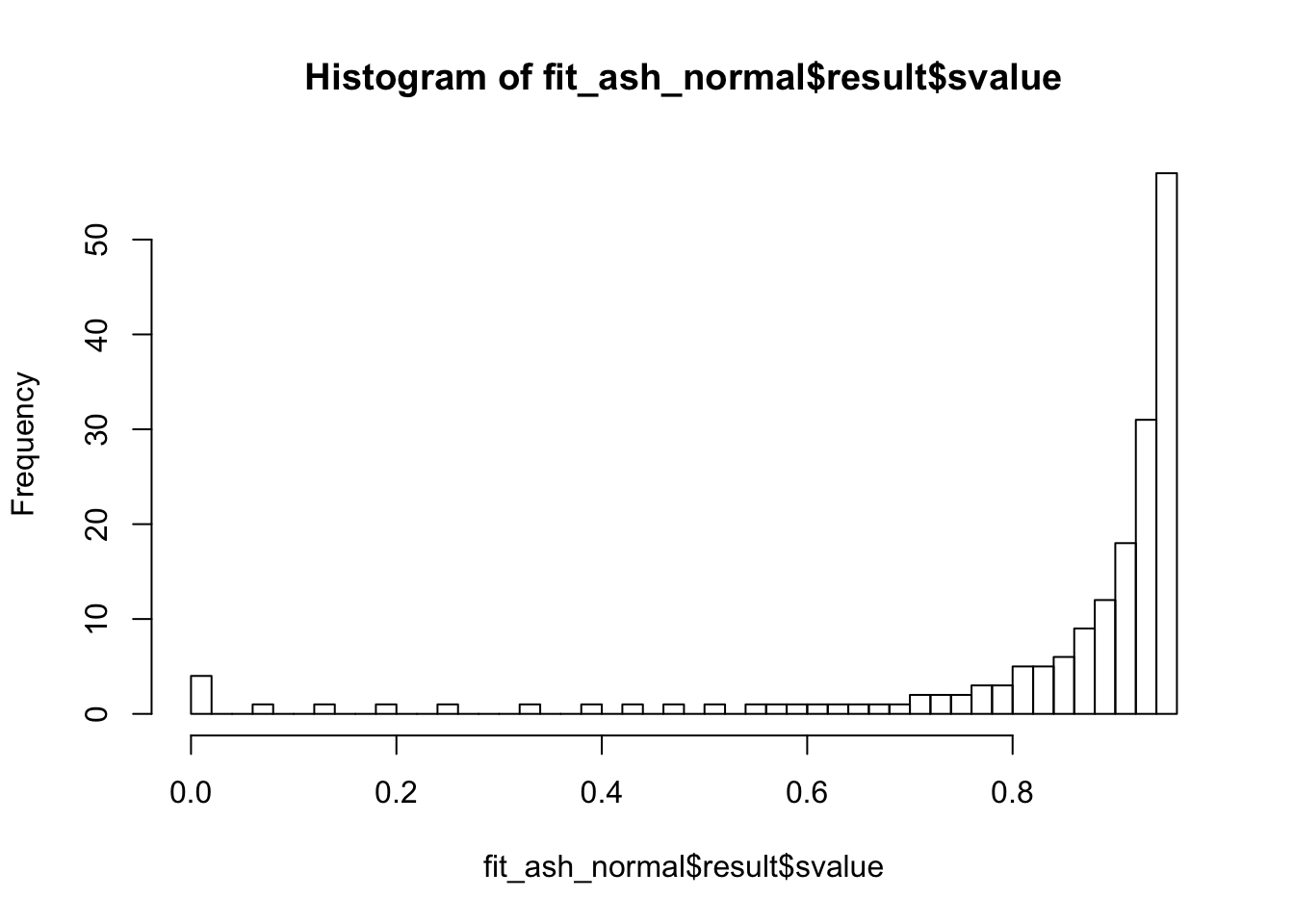
Liver specific
heart_spec <- union(union(rownames(HvC_Heart_fit_all_5perc), rownames(HvC_Kidney_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Liver_fit_all_5perc$genes, heart_spec)
X <- c(0,1,0,0,0,1,0,0,0,1,0,0,0,1,0)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 514 31
dim(methylation_values_only)
## [1] 514 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
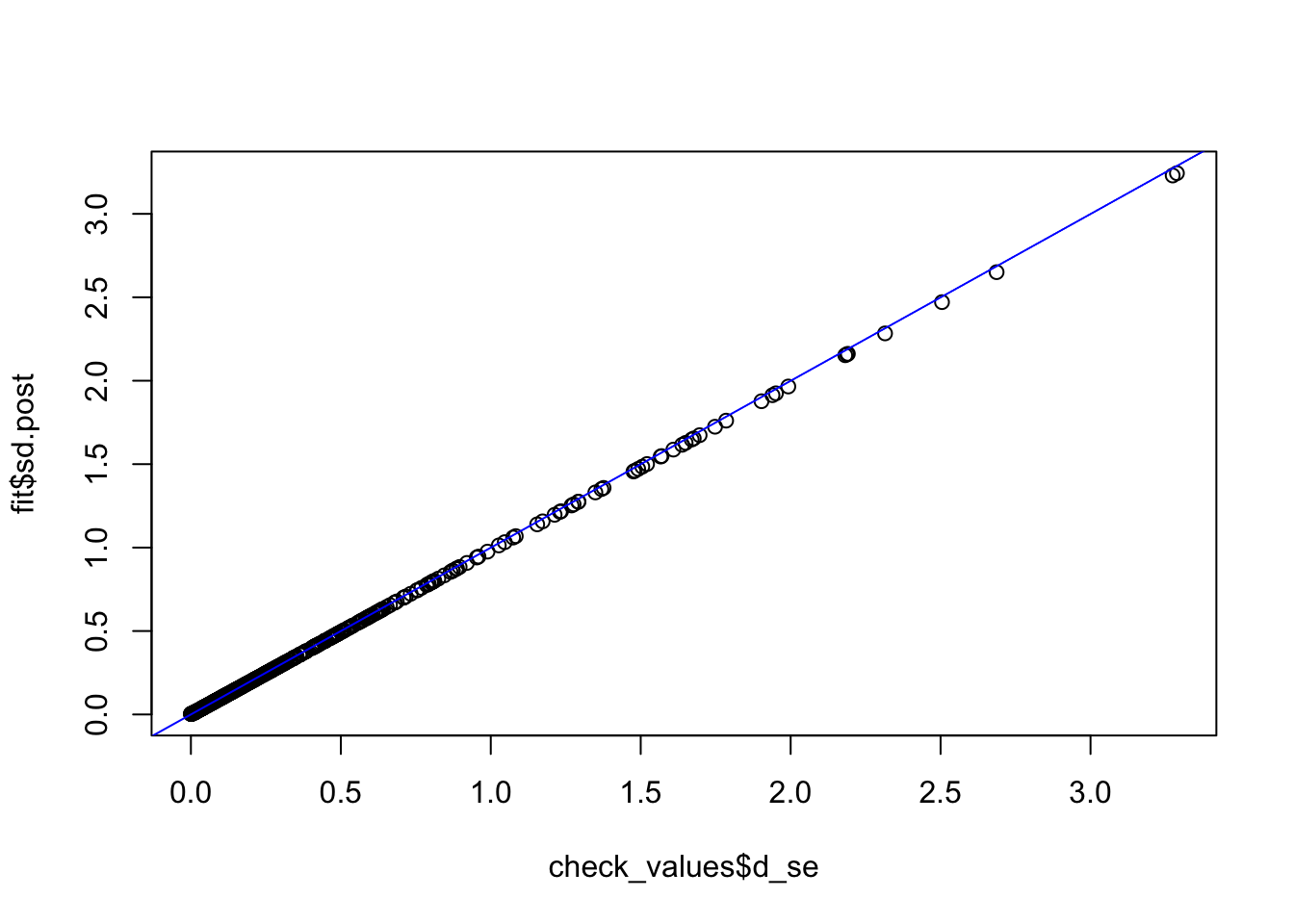
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE TRUE
## logical 512 2
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE TRUE
## logical 509 5
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 507 7
hist(fit_ash_normal$result$svalue, breaks = 50)
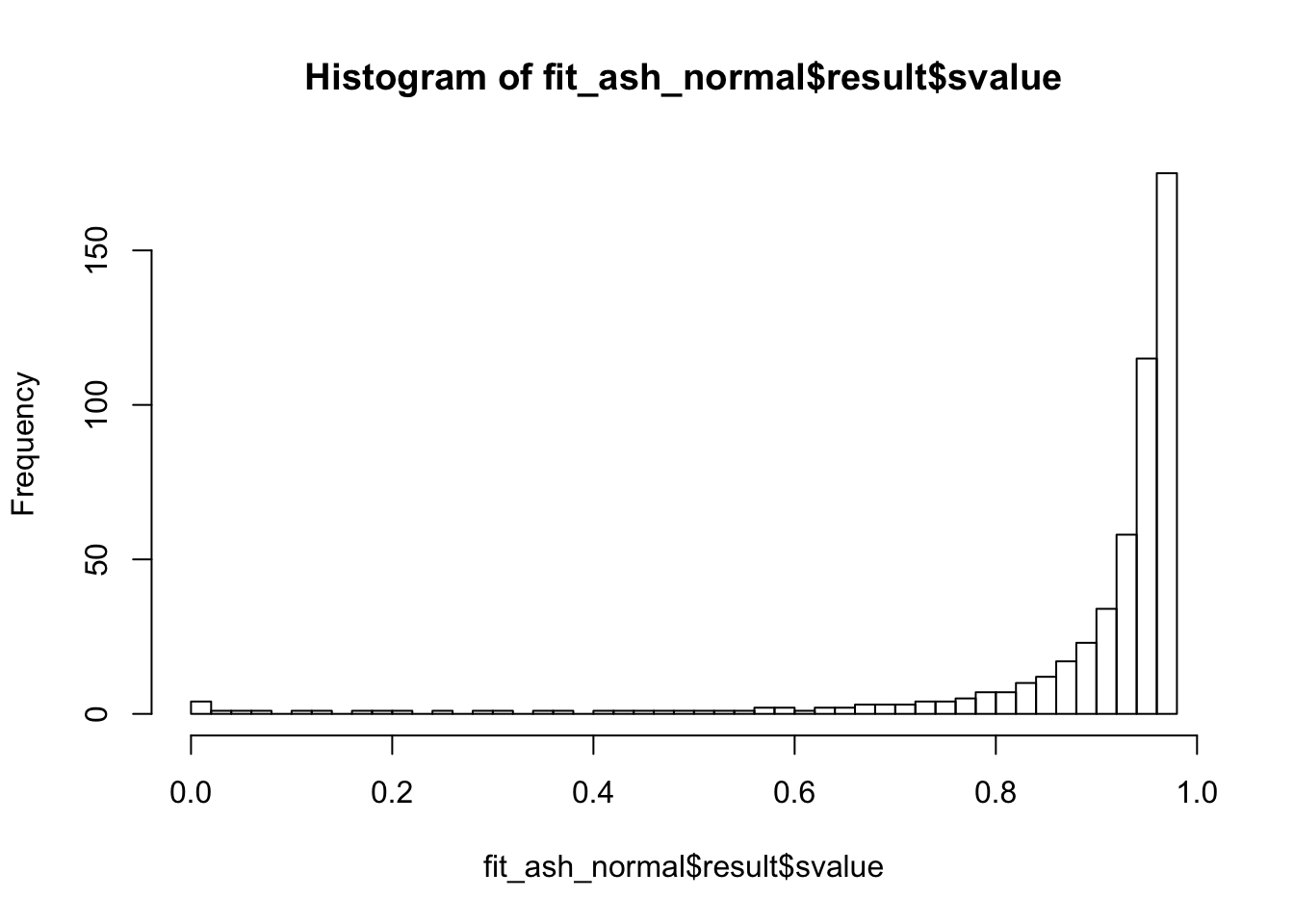
Lung specific
heart_spec <- union(union(rownames(HvC_Liver_fit_all_5perc), rownames(HvC_Heart_fit_all_5perc)), rownames(HvC_Kidney_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Lung_fit_all_5perc$genes, heart_spec_only)
X <- c(0,0,1,0,0,0,1,0,0,0,1,0,0,0,1)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 282 31
dim(methylation_values_only)
## [1] 282 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
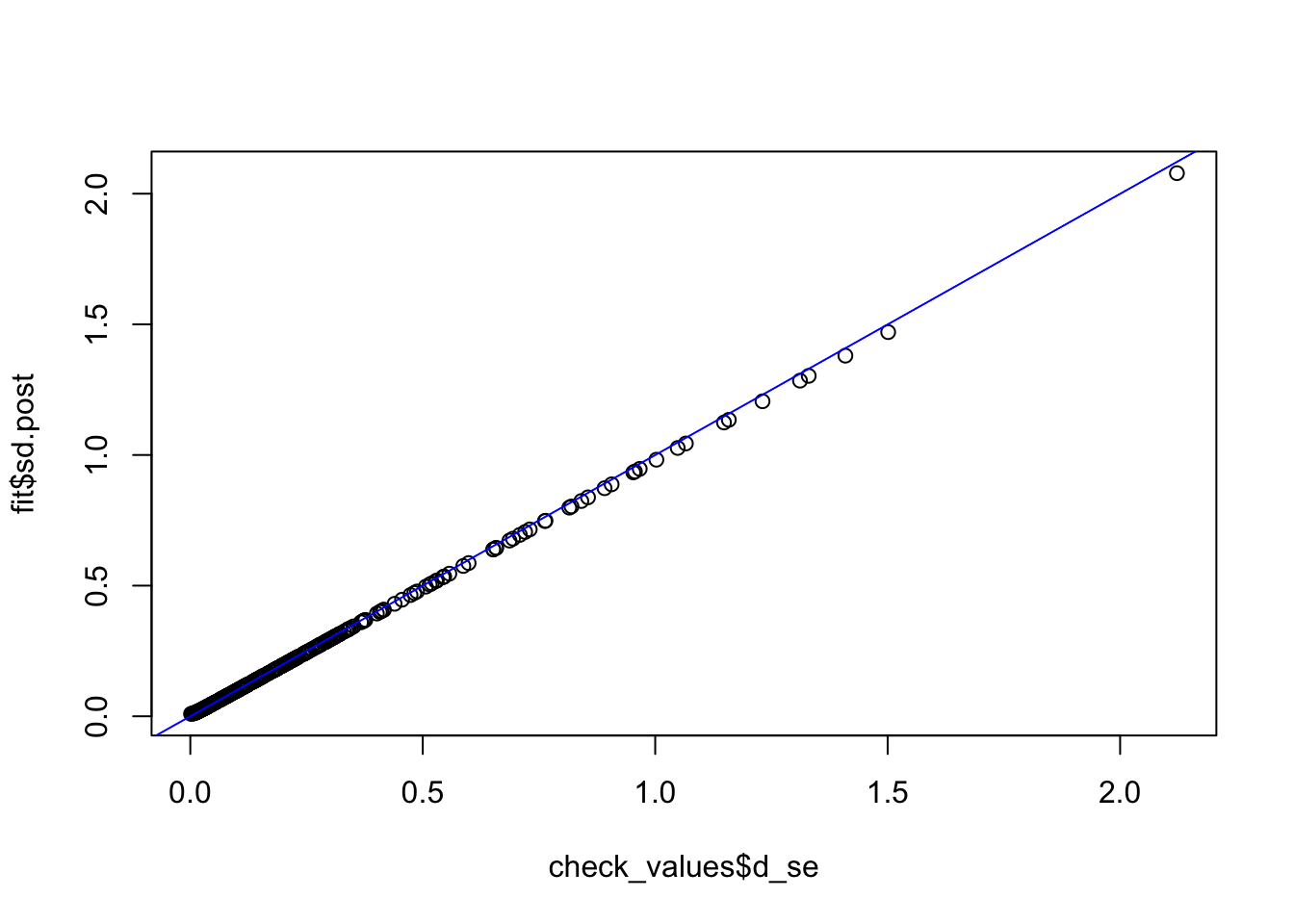
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE
## logical 282
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE
## logical 282
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE
## logical 282
hist(fit_ash_normal$result$svalue, breaks = 50)
# import sample labels
samples <- read.delim("../../../Reg_Evo_Primates/data/Sample_info_RNAseq_RIN.txt")
RIN <- samples$RIN
# expression
exprs <- read.table("../../../Reg_Evo_Primates/data/human_chimp_orth_exp_methyl_7725_hum.txt", sep="")
# methylation data
methyl <- read.csv("../../../Reg_Evo_Primates/data/chimp_human_orth_7725_avg_methyl_per_ts_gene.txt", sep="")
# Normalized gene expression data
cpm.voom.cyclic <- readRDS("../../../Reg_Evo_Primates/data/human_chimp_orth_cpm_voom_cyclic.rds")
Run the linear model (FDR level = 0.01)
human_rhesus_heart <- c(17:31)
FDR_level <- 0.01
X <- c(0,0,0,1,0,0,0,1,0,0,0,1,0,0,0)
RIN_subset <- RIN[human_rhesus_heart]
# Run for hearts
# Run the linear model in limma
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Heart_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Heart_fit_all_5perc <-
HvC_Heart_fit_all[which(HvC_Heart_fit_all$adj.P.Val < FDR_level), ]
# Run for kidneys
X <- c(1,0,0,0,1,0,0,0,1,0,0,0,1,0,0)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Kidney_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Kidney_fit_all_5perc <-
HvC_Kidney_fit_all[which(HvC_Kidney_fit_all$adj.P.Val < FDR_level), ]
# Run for livers
X <- c(0,1,0,0,0,1,0,0,0,1,0,0,0,1,0)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Liver_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Liver_fit_all_5perc <-
HvC_Liver_fit_all[which(HvC_Liver_fit_all$adj.P.Val < FDR_level), ]
# Run for lungs
X <- c(0,0,1,0,0,0,1,0,0,0,1,0,0,0,1)
design <- model.matrix(~ as.factor(X)+RIN_subset)
fit_all <- lmFit(cpm.voom.cyclic[,human_rhesus_heart], design)
fit_all <- eBayes(fit_all)
# Get results
HvC_Lung_fit_all = topTable(fit_all, coef=2, adjust="BH", number=Inf, sort.by="none")
HvC_Lung_fit_all_5perc <- HvC_Lung_fit_all[which(HvC_Lung_fit_all$adj.P.Val < FDR_level), ]
dim(HvC_Heart_fit_all_5perc)
## [1] 628 7
dim(HvC_Kidney_fit_all_5perc)
## [1] 317 7
dim(HvC_Liver_fit_all_5perc)
## [1] 906 7
dim(HvC_Lung_fit_all_5perc)
## [1] 141 7
Heart specific
heart_spec <- union(union(rownames(HvC_Kidney_fit_all_5perc), rownames(HvC_Liver_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Heart_fit_all_5perc$genes, heart_spec)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 391 31
dim(methylation_values_only)
## [1] 391 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
X <- c(0,0,0,1,0,0,0,1,0,0,0,1,0,0,0)
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
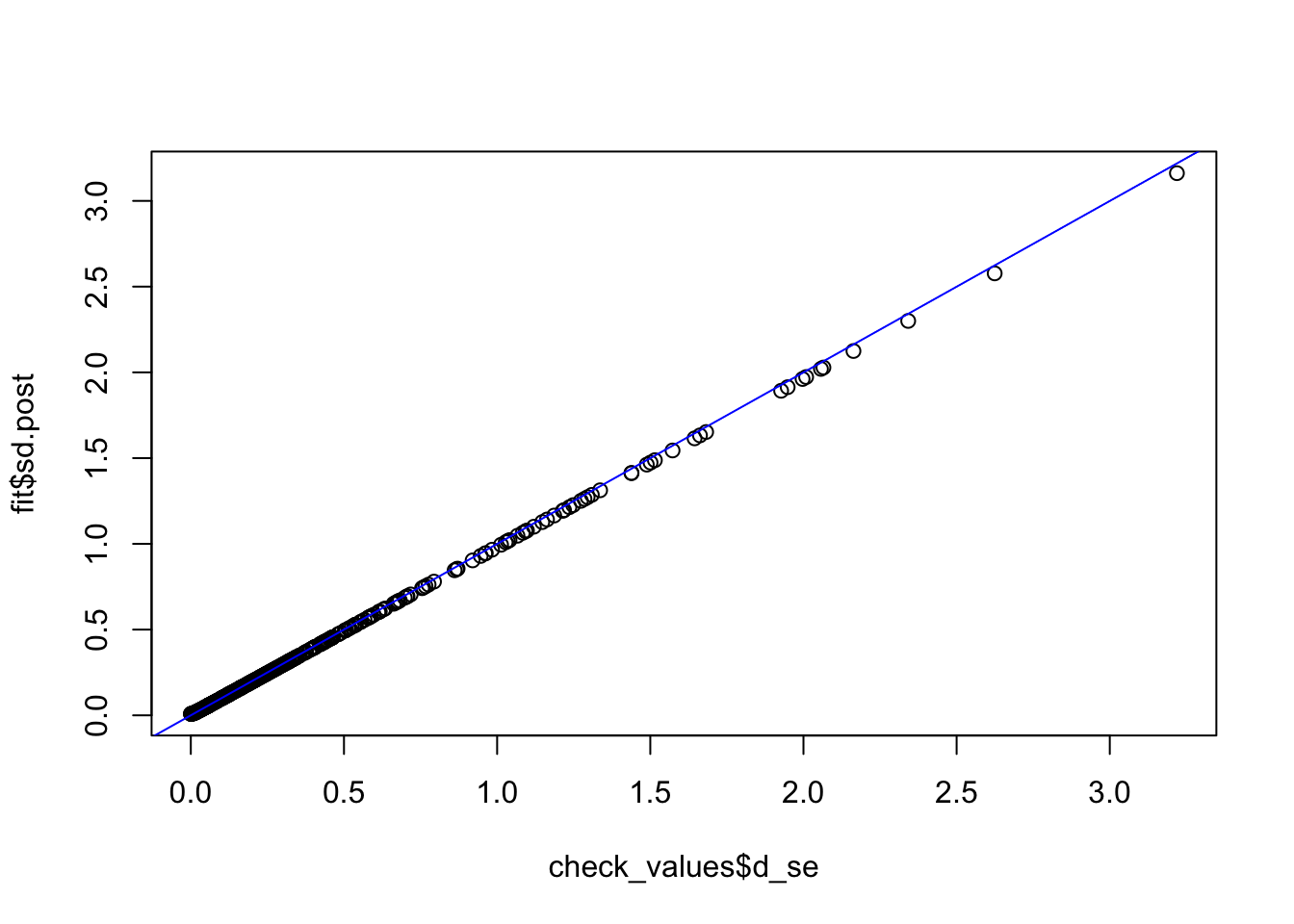
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
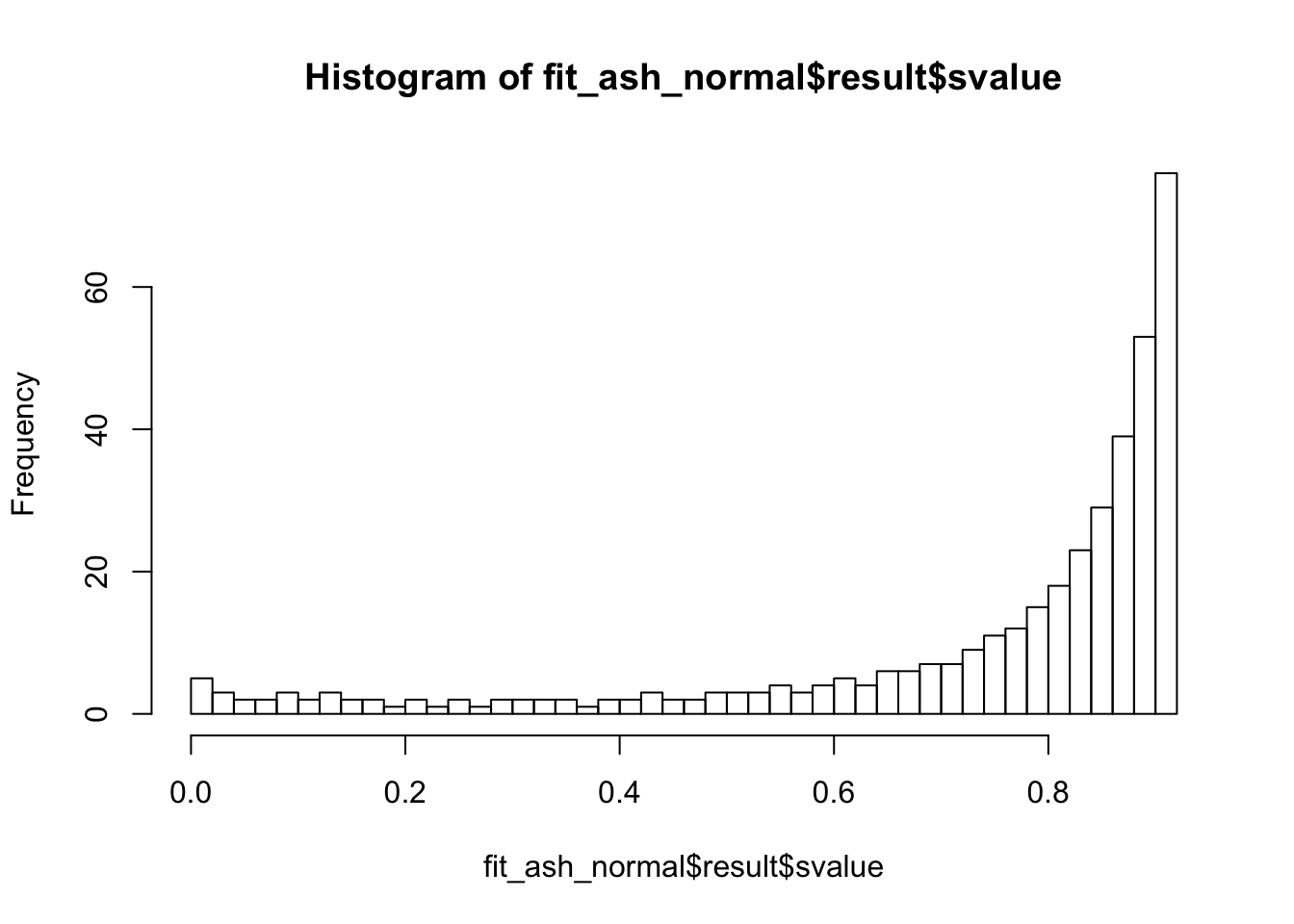
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE TRUE
## logical 389 2
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE TRUE
## logical 382 9
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 376 15
hist(fit_ash_normal$result$svalue, breaks = 50)
#which(fit_ash_normal$result$svalue < 0.1 = TRUE)
Kidney specific
heart_spec <- union(union(rownames(HvC_Heart_fit_all_5perc), rownames(HvC_Liver_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Kidney_fit_all_5perc$genes, heart_spec)
X <- c(1,0,0,0,1,0,0,0,1,0,0,0,1,0,0)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 175 31
dim(methylation_values_only)
## [1] 175 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
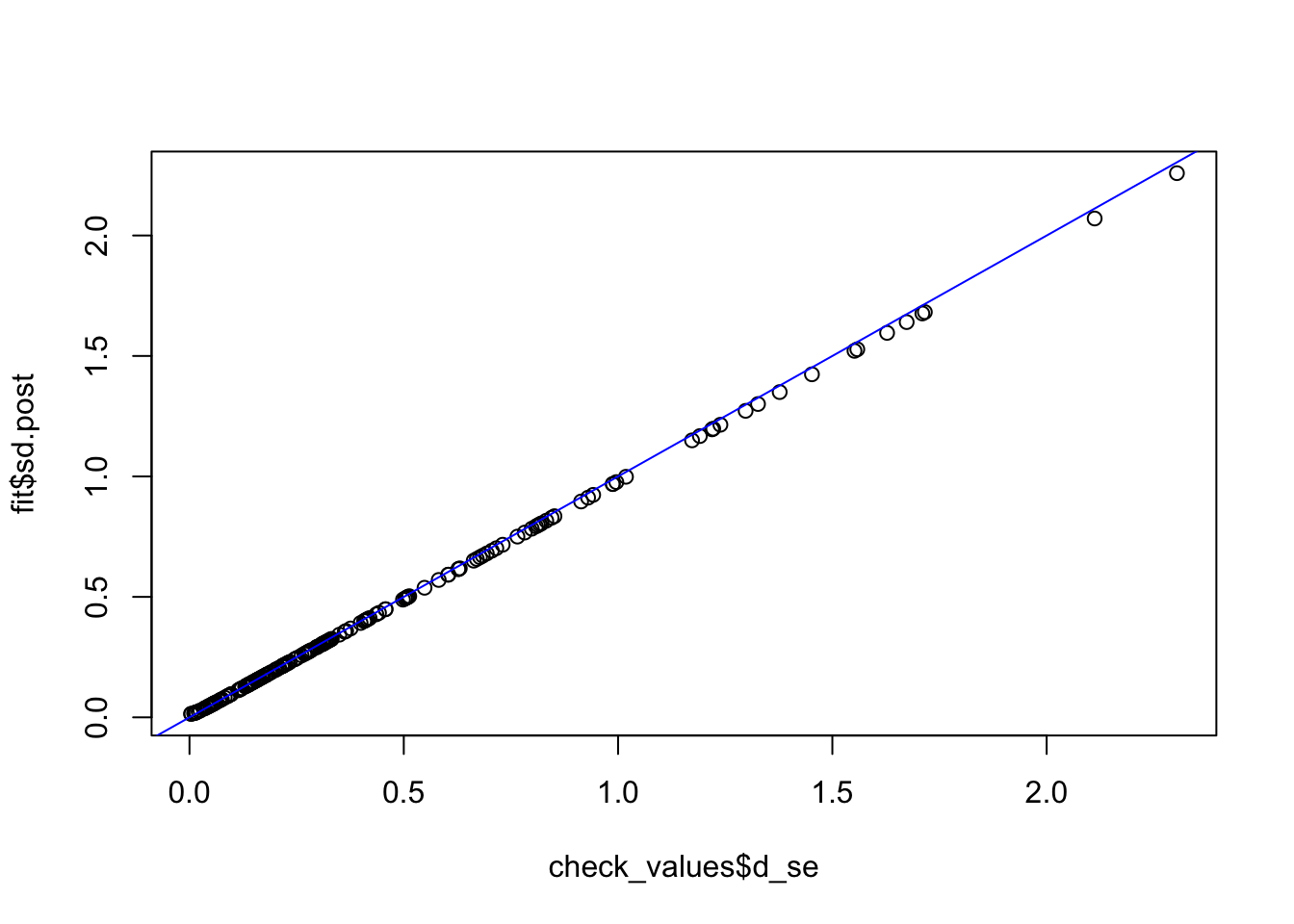
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE TRUE
## logical 167 8
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE TRUE
## logical 163 12
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 160 15
hist(fit_ash_normal$result$svalue, breaks = 50)
Liver specific
heart_spec <- union(union(rownames(HvC_Heart_fit_all_5perc), rownames(HvC_Kidney_fit_all_5perc)), rownames(HvC_Lung_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Liver_fit_all_5perc$genes, heart_spec)
X <- c(0,1,0,0,0,1,0,0,0,1,0,0,0,1,0)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 565 31
dim(methylation_values_only)
## [1] 565 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE TRUE
## logical 542 23
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE TRUE
## logical 521 44
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE TRUE
## logical 509 56
hist(fit_ash_normal$result$svalue, breaks = 50)
Lung specific
heart_spec <- union(union(rownames(HvC_Liver_fit_all_5perc), rownames(HvC_Heart_fit_all_5perc)), rownames(HvC_Kidney_fit_all_5perc))
heart_spec_only <- setdiff(HvC_Lung_fit_all_5perc$genes, heart_spec_only)
X <- c(0,0,1,0,0,0,1,0,0,0,1,0,0,0,1)
# Find which DE genes have methylation values associated with it
human_chimp_heart <- exprs[,1] %in% heart_spec_only
human_chimp_heart <- as.data.frame(human_chimp_heart)
counts_genes_in <- cbind(exprs, human_chimp_heart)
counts_genes_in_cutoff <- subset(counts_genes_in, human_chimp_heart ==
"TRUE")
# counts_genes_in_cutoff <- counts_genes_in_cutoff[,1:79]
expression_values_only <- counts_genes_in_cutoff[,2:32]
methylation_values_only <- counts_genes_in_cutoff[,49:79]
dim(expression_values_only)
## [1] 96 31
dim(methylation_values_only)
## [1] 96 31
Y <- expression_values_only[,17:31]
M <- methylation_values_only[,17:31]
check_values <- mediate.test.regressing(Y, X, M, RIN_subset)
fit <- vash(check_values$d_se,df=13, singlecomp = T, unimodal = "auto")
plot(check_values$d_se, fit$sd.post)
abline(0,1,col="blue")
fit_ash_normal <- ash(as.vector(check_values$d), fit$sd.post, mode = 0, mixcompdist = "normal")
## Due to absence of package REBayes, switching to EM algorithm
summary(fit_ash_normal$result$svalue < 0.01)
## Mode FALSE
## logical 96
summary(fit_ash_normal$result$svalue < 0.05)
## Mode FALSE
## logical 96
summary(fit_ash_normal$result$svalue < 0.1)
## Mode FALSE
## logical 96
hist(fit_ash_normal$result$svalue, breaks = 50)